
In late February, 2022 the US Food and Drug Administration (FDA) hosted a webinar detailing its current transition plans for medical devices marketed pursuant to either Emergency Use Authorization (EUA) or Enforcement Policies during the COVID-19 public health emergency (COVID-19 PHE).
The webinar sought to prepare stakeholders for the upcoming transition back to normal operations by describing its expectations for the industry over the 180-day transition period laid out in two recent draft guidance documents, Transition Plan for Medical Devices Issued EUAs During the COVID-19 PHE (EUA Transition Plan) and Transition Plan for Medical Devices That Fall Within Enforcement Policies Issued During the COVID-19 PHE (Enforcement Policy Transition Plan), which will be implemented in service to four key principles:
- an orderly, transparent transition with consistent FDA-manufacturer interactions
- a risk-based approach to different types of devices
- FDA action as necessary on a case-by-case basis and
- continued patient and provider access to devices.
Scope
Devices subject to Transition Plan requirements are listed in the draft guidances by product code; these will be updated until finalized.
The EUA Transition Plan does not apply to i) devices for which EUAs have already been, or will be, revoked because their criteria were no longer met or because other circumstances make such revocation appropriate to protect the public health or safety or ii) 564(a) good manufacturing practice deviations.
The Enforcement Policy Transition Plan does not include diagnostic tests commercialized pursuant to FDA’s Policy for Diagnostic Tests for COVID-19.
Transition plan timelines
FDA’s action recommendations for the return to normal operations follow a 180-day, three-phased approach:
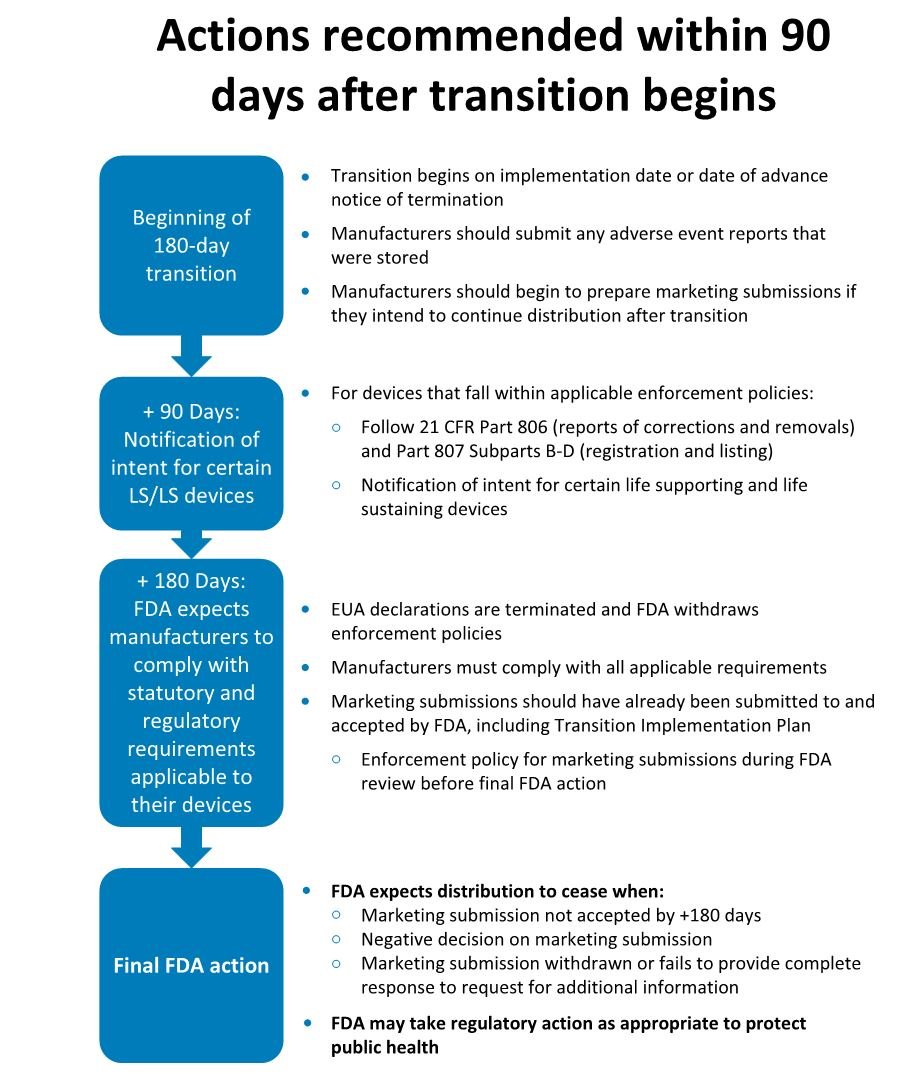
Where a manufacturer does not intend to continue distribution of COVID-19 related medical devices, those devices already in distribution at the end of the 180-day transition period may continue to be sold under the following conditions:

Scenarios not addressed in draft guidances
The timeframes and actions described in the Transition Plan apply generally. Should individual scenarios not be addressed in the guidances, FDA encourages individual manufacturers to reach out to discuss specific devices, timelines and 510(k)s, de novos and PMAs submissions, so the agency can facilitate as necessary.
FDA soliciting comments on draft guidances
FDA has invited stakeholders submit comments to the draft guidance documents by March 23, 2022 for consideration as the agency drafts the final versions.

/Passle/596df0e43d947619ec025f26/MediaLibrary/Images/2024-10-08-10-50-28-575-67050e74c71f57809f6d0411.jpg)
/Passle/596df0e43d947619ec025f26/SearchServiceImages/2025-03-17-10-33-28-399-67d7fa78bf14cca0f5e2c402.jpg)
/Passle/596df0e43d947619ec025f26/SearchServiceImages/2025-02-05-11-45-56-829-67a34f74966bf3c993454c6a.jpg)
/Passle/596df0e43d947619ec025f26/MediaLibrary/Document/2025-02-04-16-21-29-274-A27003_Life_Sciences_January_2025_Brochure_English_V1_afterchange_thumbnail_1.png)
/Passle/596df0e43d947619ec025f26/SearchServiceImages/2025-01-24-09-38-32-700-67935f98312d8a93f10c9c08.jpg)






















